

Request a demo specialized to your need.
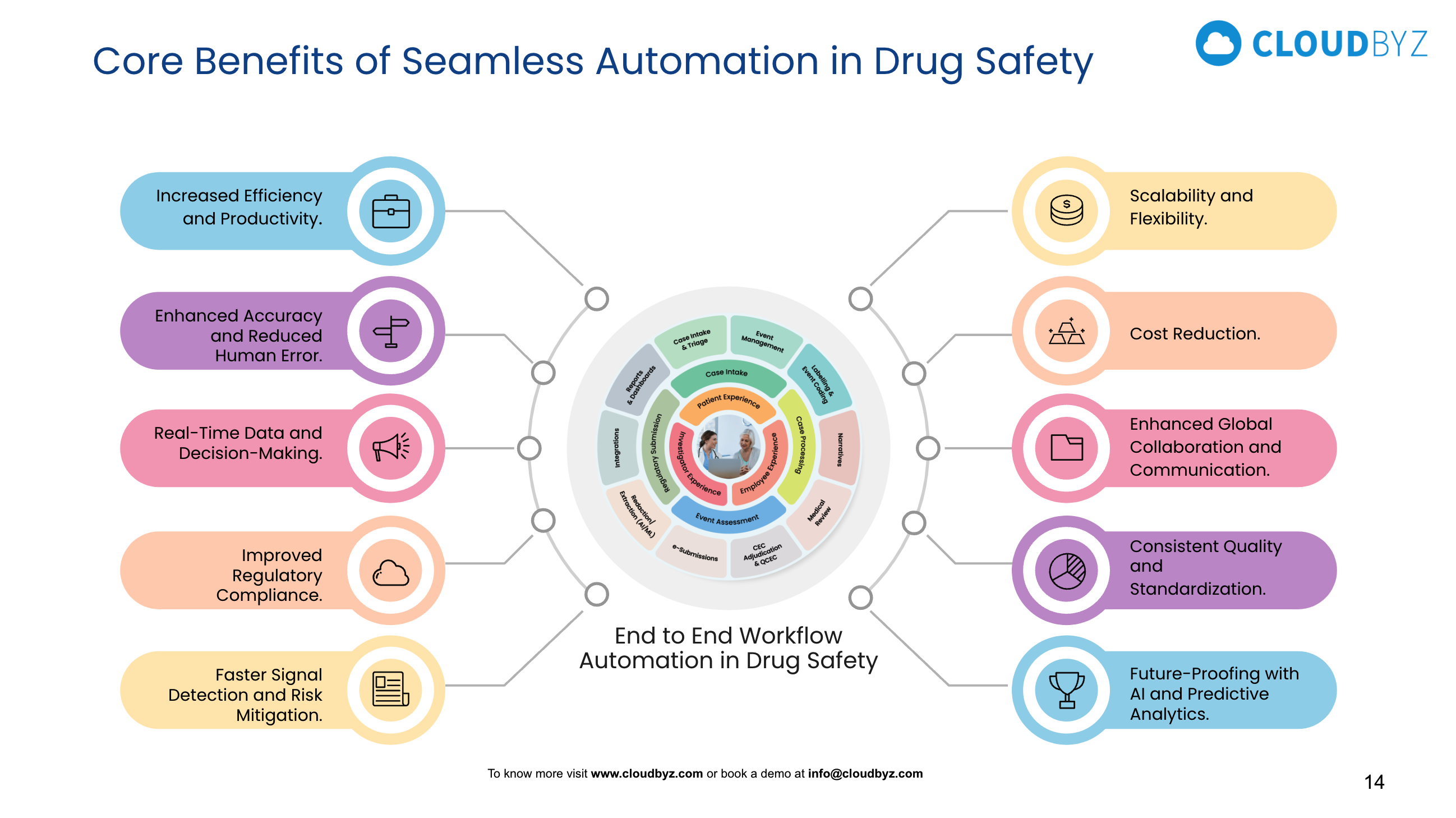
Introduction: Why Terminology Matters in Drug Safety
In pharmacovigilance, precision of language is not a bureaucratic formality — it is a patient safety imperative. A misclassified adverse event, an ambiguously coded reaction, or an incorrectly assessed causal relationship can distort signal detection, mislead regulatory decisions, and ultimately delay the identification of a genuine drug safety risk. The definitions and terminology that underpin pharmacovigilance are, therefore, the load-bearing architecture of the entire drug safety enterprise.
Yet for many practitioners — from pharmacists and clinical investigators to regulatory affairs professionals and medical monitors — the terminology can feel like an impenetrable thicket of acronyms, overlapping definitions, and jurisdictional nuance. This article cuts through that complexity, providing both a rigorous grounding in foundational pharmacovigilance concepts and a forward-looking perspective on how these definitions are evolving in an era of real-world data, artificial intelligence, and increasingly complex therapeutic modalities.
The Regulatory Foundation: What is Pharmacovigilance?
The World Health Organization (WHO) defines pharmacovigilance as "the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other medicine-related problem." This deceptively simple definition encompasses a vast ecosystem of data collection, signal management, risk communication, and regulatory interaction that spans the entire lifecycle of a medicinal product — from first-in-human clinical trials to post-market surveillance decades after approval.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has done more than any other body to standardize pharmacovigilance terminology globally. Guidelines such as ICH E2A, E2B, E2C, and E2E form the bedrock of how adverse events are defined, collected, and reported across regulatory jurisdictions including the FDA (United States), EMA (European Union), PMDA (Japan), and beyond.
Understanding pharmacovigilance begins with mastering its vocabulary.
Adverse Drug Reaction (ADR): More Than Just a Side Effect
The term Adverse Drug Reaction (ADR) is one of the most fundamental — and most frequently misunderstood — concepts in drug safety. Colloquially used interchangeably with "side effect," the ADR has a precise regulatory and clinical meaning that carries significant consequences for how events are reported and analyzed.
The Classic Definition
The WHO's original definition, formalized in 1972, describes an ADR as: "A response to a drug which is noxious and unintended, and which occurs at doses normally used in man for the prophylaxis, diagnosis, or therapy of disease, or for the modifications of physiological function."
The critical phrase here is "at doses normally used." This distinguishes an ADR from events arising from overdose, abuse, or medication error — though contemporary pharmacovigilance increasingly captures all of these in its broader net.
The Modern Evolution: ICH E2A Perspective
ICH E2A refined ADR terminology for clinical trial contexts, drawing a key distinction between Adverse Events (AEs) and ADRs:
- An Adverse Event (AE) is any untoward medical occurrence in a patient administered a medicinal product, regardless of whether there is a causal relationship to the treatment. It is a broad, inclusive term used primarily in clinical trial safety data collection.
- An ADR, by contrast, implies at minimum a reasonable possibility of a causal relationship between the drug and the event. This causal element is the defining differentiator.
This distinction is not semantic hairsplitting. It directly affects expedited reporting obligations, labeling decisions, and the risk-benefit assessments that regulators and manufacturers conduct throughout a product's lifecycle.
Spontaneous ADR Reports and the Pharmacovigilance Database
In the post-market setting, ADR reports are predominantly collected through spontaneous reporting systems — the FDA's FAERS (FDA Adverse Event Reporting System), the EMA's EudraVigilance, and the WHO's VigiBase, which holds over 30 million individual case safety reports from more than 130 member countries. These databases form the epidemiological backbone of global signal detection, and their integrity depends entirely on consistent application of ADR definitions.
Serious Adverse Events (SAEs): The Threshold for Urgency
If the ADR defines what is captured, the concept of Serious Adverse Events (SAEs) defines which events demand immediate action. The seriousness criteria, established in ICH E2A, represent a regulatory bright line that triggers expedited reporting timelines — typically 7 or 15 calendar days depending on jurisdiction and expectedness.
ICH E2A Seriousness Criteria
An adverse event or adverse drug reaction is considered serious if it meets one or more of the following criteria:
- Fatal — results in death
- Life-threatening — places the patient at immediate risk of death at the time of the event
- Requires inpatient hospitalization or prolongation of existing hospitalization
- Results in persistent or significant disability or incapacity
- Is a congenital anomaly or birth defect
- Is an important medical event — a catch-all category requiring medical judgment, covering events that may not immediately meet the above criteria but may jeopardize the patient or require medical/surgical intervention to prevent one of the above outcomes
The Serious vs. Severe Distinction: A Critical Nuance
One of the most common errors in pharmacovigilance practice is conflating serious with severe. These are independent dimensions of an adverse event:
- Severity refers to the intensity of an event — mild, moderate, or severe — on a clinical spectrum.
- Seriousness is a regulatory classification based on the outcome criteria above.
A severe headache, for example, may be clinically debilitating but not serious by regulatory definition if it does not result in hospitalization or any of the other seriousness criteria. Conversely, a mild rash accompanied by anaphylactic potential may qualify as an important medical event, rendering it serious despite being clinically mild at presentation. Regulatory professionals who internalize this distinction make significantly more defensible safety reporting decisions.
SAEs in Clinical Trials vs. Post-Market Settings
In clinical trials, SAE reporting is governed by strict contractual and regulatory obligations — sponsors must receive and assess SAE reports from investigators within defined timelines, and those that are also unexpected and possibly related to the investigational product (i.e., SUSARs — Suspected Unexpected Serious Adverse Reactions) must be expedited to regulators. In the post-market setting, the same seriousness criteria apply but reporting workflows differ by jurisdiction, product type, and whether the event was solicited or spontaneous.
MedDRA: The Global Medical Dictionary Powering Safety Data
Collecting an adverse event report is only half the challenge. The other half is coding it — translating free-text clinical narratives into standardized terminology that enables consistent aggregation, analysis, and signal detection across millions of reports. This is where MedDRA (Medical Dictionary for Regulatory Activities) becomes indispensable.
What is MedDRA?
MedDRA is a clinically validated, internationally standardized medical terminology developed under the auspices of ICH and maintained by the MSSO (MedDRA Maintenance and Support Services Organization). First released in 1999, it is now the mandated coding dictionary for adverse event reporting to most major regulatory authorities worldwide, including the FDA, EMA, PMDA, and Health Canada.
The Five-Level Hierarchy
MedDRA's architecture is a hierarchical structure with five levels, each providing a different degree of specificity and analytical utility:
- System Organ Class (SOC) — The broadest level, organizing terms by etiology, anatomical location, or purpose (e.g., Cardiac disorders, Infections and infestations). There are 27 SOCs in MedDRA.
- High Level Group Term (HLGT) — A superordinate grouping within an SOC, providing an intermediate layer of organization (e.g., Cardiac arrhythmias within Cardiac disorders).
- High Level Term (HLT) — A further refinement within an HLGT, grouping related Preferred Terms (e.g., Rate and rhythm disorders NEC).
- Preferred Term (PT) — The primary level used for regulatory reporting. Each PT represents a distinct medical concept with a unique eight-digit code (e.g., Ventricular tachycardia, PT code 10047302). This is the level at which individual case safety reports are coded.
- Lowest Level Term (LLT) — The most granular level, designed to capture the precise language used by reporters. LLTs map upward to PTs and are the entry point for case coding (e.g., Fast ventricular rhythm maps to the PT Ventricular tachycardia).
Standardized MedDRA Queries (SMQs)
A major analytical tool within MedDRA is the Standardized MedDRA Query (SMQ) — validated groupings of MedDRA terms related to a defined medical condition or area of interest. SMQs allow safety teams to cast a wide analytical net when screening databases for signals related to complex clinical entities (such as Drug-induced liver injury, Stevens-Johnson Syndrome, or Embolic and thrombotic events) that may be coded across many different PTs.
The Art and Science of MedDRA Coding
Effective MedDRA coding requires both technical mastery and clinical judgment. The guiding principle — code the diagnosis when known, rather than signs and symptoms — sounds straightforward but demands nuanced application. A report describing "elevated liver enzymes, jaundice, and right upper quadrant pain" should ideally be coded to Hepatocellular injury or Drug-induced liver injury rather than three separate symptom PTs, if the diagnosis is established. However, when a diagnosis has not been confirmed, coding at the symptom level may be more accurate and less assumptive.
MedDRA is updated twice annually (March and September), with new terms added and retired terms flagged — requiring ongoing maintenance of legacy coded datasets and pharmacovigilance systems. The dictionary currently contains over 80,000 terms across all levels, reflecting the complexity of modern clinical medicine.
Causality Assessment: The Intellectual Heart of Pharmacovigilance
Of all the cognitive tasks in drug safety, causality assessment — determining whether a drug caused or contributed to an observed adverse event — is the most intellectually demanding and the most consequential. It is where clinical medicine, epidemiology, pharmacology, and regulatory science converge.
Why Causality is Difficult
The fundamental challenge is counterfactual: we cannot know with certainty what would have happened to a patient had they not taken the drug. Adverse events often arise in populations who are already ill, taking multiple medications, and subject to the natural history of their underlying conditions. Establishing that this drug caused this event in this patient requires systematic reasoning under uncertainty.
The WHO-Uppsala Monitoring Centre (WHO-UMC) Causality Categories
The most widely used framework for individual case causality assessment is the WHO-UMC system, which categorizes the probability of a causal relationship between a drug and an event into six levels:
- Certain — The event follows a plausible time sequence; it responds to withdrawal (dechallenge); it recurs on re-administration (rechallenge); it cannot be explained by the patient's clinical state, other drugs, or chemicals; and the event is pharmacologically plausible or documented definitively.
- Probable/Likely — The event follows a reasonable time sequence; it is unlikely to be attributed to the disease or other drugs; the response to withdrawal is clinically reasonable; rechallenge information is not required.
- Possible — The event follows a reasonable time sequence; it could also be explained by the disease or other drugs; information on drug withdrawal may be lacking or unclear.
- Unlikely — The temporal association is improbable; the event could be explained by disease or other drugs.
- Conditional/Unclassified — The event requires more data for proper assessment, or additional data are being analyzed.
- Unassessable/Unclassifiable — A report suggesting an adverse reaction that cannot be judged because information is insufficient or contradictory.
The Naranjo Algorithm: A Structured Scoring Approach
The Naranjo Adverse Drug Reaction Probability Scale, developed in 1981 by Naranjo and colleagues, provides a structured, quantitative approach to causality assessment through a 10-question scoring system. Questions address temporal relationship, known pharmacological plausibility, dechallenge and rechallenge outcomes, alternative explanations, and objective confirmation. Scores are interpreted as:
- ≥9: Definite ADR
- 5–8: Probable ADR
- 1–4: Possible ADR
- ≤0: Doubtful ADR
While the Naranjo scale is widely used in clinical and academic settings, it has limitations — particularly its relative insensitivity to complex polypharmacy scenarios and its dependence on rechallenge data that is often ethically unavailable.
The Bradford Hill Criteria: Population-Level Causality
At the aggregate level, pharmacovigilance scientists frequently invoke the Bradford Hill criteria — originally developed for epidemiological causal inference — to evaluate whether a statistical signal from a safety database represents a true causal relationship. The nine criteria include strength of association, consistency across studies, specificity, temporality, biological gradient, plausibility, coherence, experimental evidence, and analogy. No single criterion is either necessary or sufficient, but convergent evidence across multiple criteria builds a compelling causal argument.
Expectedness: A Related but Distinct Concept
Closely related to causality is the concept of expectedness — whether the nature, severity, or frequency of an adverse reaction is consistent with the current product labeling (the Reference Safety Information, or RSI). A reaction is unexpected if it is not listed in the RSI or if it occurs at a severity, specificity, or frequency beyond what is described. Expectedness, combined with causality and seriousness, determines expedited reporting obligations and triggers label update discussions.
Emerging Challenges: Where Terminology Must Evolve
Real-World Data and Digital Health
The proliferation of electronic health records, claims databases, patient registries, and wearable health technologies has created vast new streams of pharmacovigilance-relevant data. However, these data sources were not designed for safety surveillance — they use heterogeneous clinical terminologies (ICD-10, SNOMED-CT, CPT) that require mapping to MedDRA, introducing noise and inconsistency. The field must develop robust, validated methodologies for translating real-world data into actionable pharmacovigilance insights without losing the definitional precision that regulatory decision-making demands.
Artificial Intelligence and Automated Case Processing
Machine learning tools are increasingly being applied to automate case triage, MedDRA coding, and even causality assessment. While these tools offer significant efficiency gains — particularly in managing the exponential growth in Individual Case Safety Reports (ICSRs) — they also risk encoding definitional inconsistencies at scale. A poorly trained coding algorithm that systematically miscodes a class of reactions can corrupt signal detection for entire product categories. Regulatory agencies including the FDA and EMA are actively developing frameworks for the validation and oversight of AI-driven pharmacovigilance tools.
Gene Therapies, Cell Therapies, and ATMPs
Advanced Therapy Medicinal Products (ATMPs) — including gene therapies, CAR-T therapies, and tissue-engineered products — present definitional challenges that existing pharmacovigilance frameworks were not designed to address. When a gene therapy integrates into a patient's genome and produces effects years later, conventional notions of temporal association and dechallenge become conceptually incoherent. The field is actively grappling with how to adapt ADR definitions and causality assessment frameworks for modalities with indefinite pharmacological persistence.
Patient-Reported Outcomes and Consumer Terminology
An increasing proportion of safety data originates directly from patients — through social media monitoring, mobile health applications, and direct patient reporting portals. Patients describe their experiences in natural language far removed from MedDRA's clinical terminology. Bridging consumer vernacular ("my heart was racing and I felt like I was going to pass out") with standardized medical coding without losing clinical meaning is a growing area of innovation in natural language processing and pharmacovigilance informatics.
Conclusion: Mastery of Terminology as Professional Responsibility
Pharmacovigilance is ultimately a discipline of language as much as it is of science. The definitions of ADR, SAE, and the hierarchical structure of MedDRA are not abstract regulatory constructs — they are the instruments through which patient safety signals are captured, communicated, and acted upon. Causality assessment frameworks are the intellectual tools through which we distinguish drug-induced harm from the noise of coincidental events in sick populations.
For practitioners in drug safety, regulatory affairs, clinical development, and medical affairs, mastery of this terminology is not optional — it is a core professional competency. The decision to classify an event as serious or non-serious, expected or unexpected, possibly or probably related, can determine whether a safety signal is flagged in time to prevent harm to future patients.
As the landscape of therapeutics grows more complex and the volume of safety data continues to expand, the foundational definitions of pharmacovigilance must be both rigorously preserved and thoughtfully evolved. The stakes — measured in patient lives — demand nothing less.
Subscribe to our Newsletter
