

Request a demo specialized to your need.
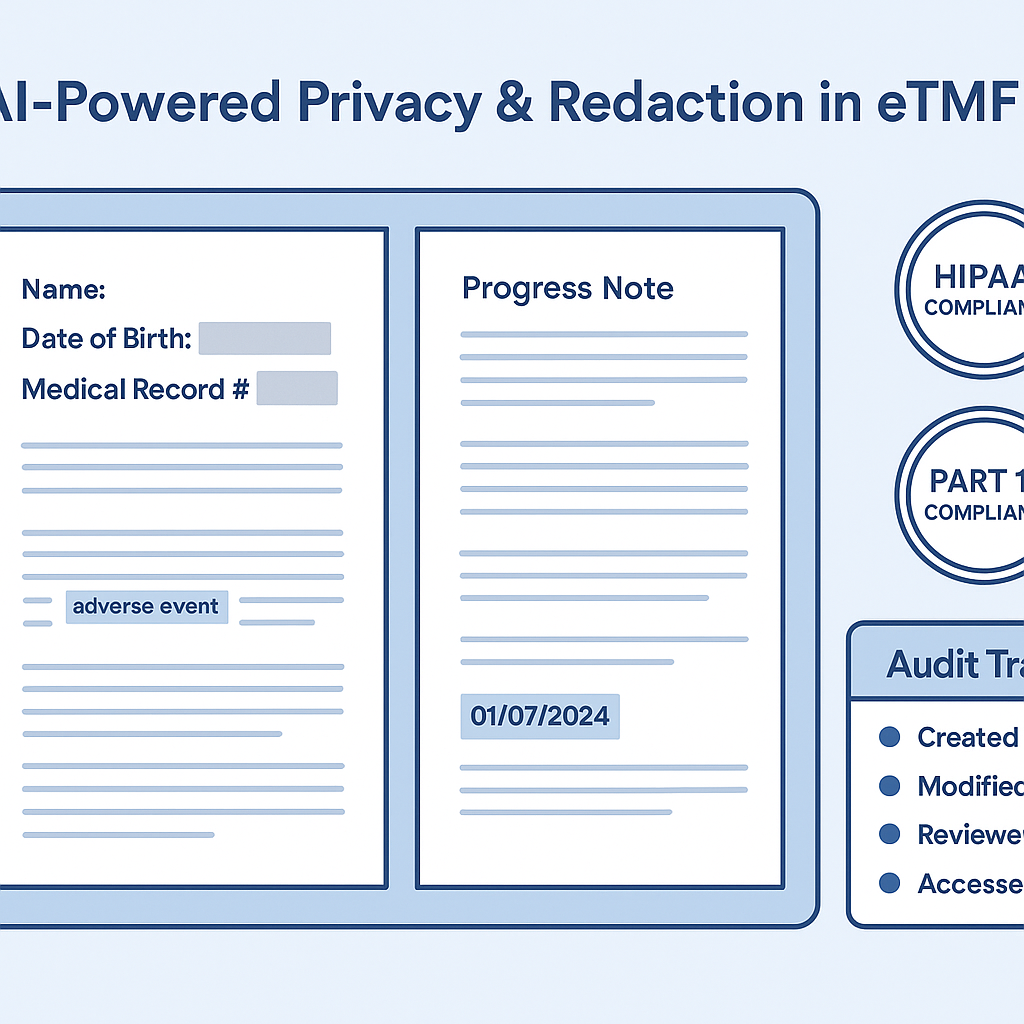
Blueprint for explainable AI redaction that protects PHI in eTMF.
The electronic Trial Master File (eTMF) sits at the center of clinical trial compliance, audit readiness, and regulatory trust. It is also one of the largest repositories of sensitive information in life sciences—containing patient identifiers, investigator details, site contracts, signatures, and regulated personal data that must be protected across jurisdictions.
As trials become more global, decentralized, and data-intensive, privacy risk in the eTMF has grown exponentially. Manual redaction processes—once considered sufficient—are now a source of delay, inconsistency, and regulatory exposure. AI-driven redaction represents a critical evolution: moving eTMF privacy from a reactive, human-dependent activity to an intelligent, scalable, and auditable control embedded directly into document workflows.
The Privacy Challenge Inside the eTMF
The eTMF was never designed to be privacy-neutral. It aggregates documents from multiple sources, formats, and stakeholders, including scanned PDFs, handwritten notes, structured reports, and correspondence. These documents routinely contain:
-
Direct patient identifiers (names, initials, dates of birth, IDs)
-
Indirect identifiers (site numbers, rare disease references)
-
Investigator and site PII
-
Signatures, credentials, and contact details
-
Region-specific personal data protected by GDPR, HIPAA, and other regulations
Traditional redaction methods rely on manual review or basic keyword search, both of which fail under scale. Human reviewers miss context, tire over time, and struggle with unstructured or scanned content. Keyword-based tools over-redact or under-redact, creating operational inefficiency and compliance risk.
Why Manual Redaction No Longer Scales
Manual redaction introduces three systemic risks:
Inconsistency – Different reviewers interpret redaction rules differently, leading to uneven application across countries, studies, and inspection artifacts.
Latency – Redaction becomes a bottleneck for submissions, inspections, and TMF completeness, particularly when documents must be re-reviewed after amendments or health authority requests.
Audit Exposure – Regulators increasingly expect demonstrable, repeatable privacy controls. Manual processes are difficult to defend when asked how privacy was consistently enforced.
In short, privacy cannot remain a human-only control in an AI-scale document ecosystem.
What Is AI Redaction for eTMF?
AI redaction applies machine learning, natural language processing (NLP), and computer vision to automatically detect, classify, and redact sensitive content within eTMF documents—while preserving document usability, structure, and auditability.
Crucially, AI redaction is not just about masking text. It is about understanding context, regulatory intent, and inspection use cases.
Core Capabilities of AI-Driven eTMF Redaction
Context-Aware Entity Detection
Unlike simple pattern matching, AI models understand context. They distinguish between:
-
A subject ID used as a coded reference (often allowed)
-
A subject name embedded in narrative text (must be redacted)
-
Investigator names that may be permissible internally but restricted externally
This contextual intelligence dramatically reduces false positives and false negatives.
Multi-Modal Document Intelligence
eTMF content is rarely clean or structured. AI redaction engines operate across:
-
Native PDFs and Word documents
-
Scanned images and handwritten notes (via OCR + vision models)
-
Tables, headers, footers, and embedded metadata
This ensures privacy controls are applied consistently, regardless of document format.
Rule-Driven, Jurisdiction-Aware Redaction
Privacy rules vary by region and use case. AI redaction systems support configurable policies that align with:
-
GDPR vs HIPAA requirements
-
Internal TMF access vs external inspection sharing
-
Submission-specific redaction profiles
Redaction is therefore policy-driven, not ad hoc.
Embedding AI Redaction into the eTMF Workflow
The true power of AI redaction emerges when it is embedded directly into eTMF workflows—not treated as a downstream clean-up step.
Documents can be automatically analyzed at intake, with sensitive content flagged and redacted before filing. Human reviewers operate in a human-in-the-loop model, validating AI decisions, handling edge cases, and approving redaction outcomes. Every action is logged, versioned, and traceable.
This creates a defensible privacy chain of custody from document ingestion to inspection readiness.
Auditability and Regulatory Confidence
AI redaction strengthens—not weakens—inspection readiness when designed correctly. Modern systems provide:
-
Redaction audit trails (what was redacted, why, and when)
-
Version comparisons between original and redacted documents
-
Evidence of consistent policy application
-
Reviewer oversight and electronic sign-off
When regulators ask “How do you ensure personal data is protected in your TMF?”, organizations can answer with systems, evidence, and governance—not anecdotes.
Strategic Value Beyond Compliance
AI redaction is often positioned as a compliance tool, but its strategic value is broader:
-
Faster submissions and inspection responses
-
Reduced reliance on outsourced redaction services
-
Lower risk of privacy breaches and remediation costs
-
Scalable support for decentralized and global trials
It enables privacy-by-design in clinical documentation, aligning with modern regulatory expectations and enterprise risk management.
The Future: From Redaction to Intelligent Privacy Management
AI redaction is only the beginning. The next generation of privacy intelligence will include:
-
Predictive identification of high-risk documents
-
Continuous privacy monitoring across the TMF
-
Automated privacy impact assessments
-
Explainable AI decisions aligned with regulatory guidance
As regulators increase scrutiny on data protection, organizations that embed intelligent privacy controls into their eTMF infrastructure will be better positioned to scale trials without scaling risk.
Conclusion: Privacy as a First-Class Citizen in the eTMF
AI redaction transforms eTMF privacy from a manual obligation into a systemic, intelligent capability. It ensures sensitive data is protected consistently, efficiently, and defensibly—without slowing down clinical execution.
In an environment where trust, transparency, and data protection are inseparable, AI-powered eTMF redaction is no longer optional. It is a foundational pillar of modern clinical trial governance—where compliance is built in, not bolted on.
Subscribe to our Newsletter
